|

FDA removes REMS requirements for embryofetal toxicity risk from all endothelin receptor antagonist medicines
Based on a new analysis of human pregnancy data compiled about endothelin receptor antagonist (ERA) medicines for two decades, the U.S. Food and Drug Administration (FDA) has determined that risk evaluation and mitigation strategy (REMS) requirements for embryofetal toxicity (EFT) risk are no longer necessary to ensure the benefits of ERA medicines outweigh the risks. The Agency’s current assessment is that labeling alone is adequate to communicate the risk, and labeling will continue to inform health care professionals and patients of the risk.
In April 2025, FDA eliminated the REMS for the following ERA medicines:
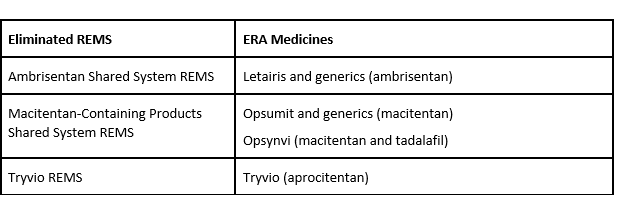 Elimination of these REMS means:
- Health care professionals can prescribe and dispense ambrisentan, macitentan-containing products, and Tryvio (aprocitentan) without enrolling or participating in a REMS.
- Patients can receive ambrisentan and macitentan-containing products without enrolling in a REMS; the Tryvio REMS did not require patient enrollment.
- Pharmacies and health care facilities can dispense ambrisentan, macitentan-containing products, and Tryvio (aprocitentan) without enrolling or participating in a REMS.
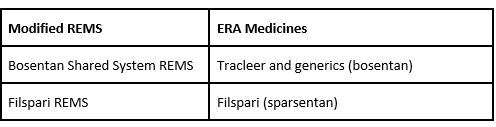
Additionally, FDA notified manufacturers for the following ERA products that they must modify the REMS to remove requirements related to EFT risk. FDA will notify prescribers, pharmacies, and patients when these REMS have been modified in fall 2025. The REMS requirements related to hepatotoxicity (liver damage) risk for these ERA medicines will remain in effect.
 Read the full update on the ERA REMS elimination.
|

